
Home
Slice Tools
Micro-assemblers
Resuable Components
Slice Tools
Michael Schatz
DNA sequencing technology is currently limited to reading upto 1000 base pairs of DNA in a given sequencing reaction. Consequently, the most commonly used process for sequencing an entire genome begins by shearing the DNA into thousands or millions of short fragments, and sequencing those short fragment known as reads. Those reads are then assembled using sophisticated assembly programs such as the Celera Assembler or AMOS. The contiguous stretches of DNA assembled in this way are called contigs. Conceptually, it is very similiar to assembling small bricks (reads) into a brick wall (contigs).
The Slice Tools are a toolkit for manipulating contigs of a genomic assembly and performing various operations on them. There are currently tools for performing basic tasks such as reverse complementing contigs, or removing reads from a contig, but there are also more sophisicated tools for joining separate contigs together via a "Micro-Assembly" operation. Unlike a full assembler, the micro-assembler operations take pre-existing contigs as an input, and new reads or other contigs are "stitched" onto them while keeping the layout of the original contig essentially unmodified.
The framework of the Slice Tools is centered around a slice view of a contig. The Slice view rotates the traditional contig view of an assembly on to its edge to shift the focus from horizontal sequences to vertical slices. The shift to a Slice view is purely a change in representation, as there is no gain or loss in information. However, depending on the operation, operations that are be difficult or cumbersome within a contig view can become extremely easy to perform.
The Slice Tools interact through the common format of Slice XML, which allows them to share a common language and a common processing core. This also allows for the tools to work in concert with the output of one tool as the input to the next in an automated pipeline similiar to how cut, sed, awk, and grep can be connected to perform more complicated manipulations on text than any single tool could.
The Slice Tools provide the mechanism to transform slices and provide the basic functionality for more complicated manipulations. The operations each tool performs tends to be very specialized and focused. For instance the tool trSlice renumber slices (translate their position in the assembly) and another tool, revSlice, calculates the reverse compliment of slices. The advantage to this system is that many small tools can quickly and easily be combined to perform novel manipulations from a common toolbox.
Even though an individual tool may only perform a single simple task, they can work in concert through the common Slice XML format to perform very complicated manipulations. For instance, by using getCoverage to break an assembly into two pieces, and then using stripSlice followed by mergeSlice, an overcollapsed repeat region can be 'pulled' apart and put into a proper assembly. This new assembly can be quickly inspected in a text editor, then converted to back to the original contig format.
Example use of combining Slice Tools to fix a collapsed repeat:
% getCoverage -x 1 -y 1100 repeat.contig > 1.slice % getCoverage -x 900 -y 2000 repeat.contig > 2.slice % stripSlice -i 5 1.slice -o 1.strip.slice % stripSlice -i 4 2.slice -o 2.strip.slice % mergeSlice 1.strip.slice 2.strip.slice | slice2contig - -o fixed.contig |
Understanding Slices
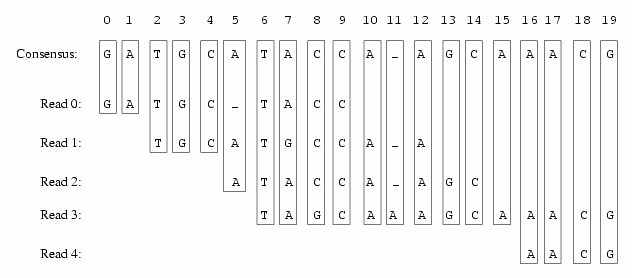
A slice is a one base wide cut of an assembly from zero or more reads. Each read in the slice contributes a base, quality value, and direction. This information can be used to compute the consensus or quality class of the slice, or the slices themselves can be manipulated to rearrange or reassemble the assembly.
If you consider the traditional contig view of an assembly to be "horizontal", meaning the assembly is oriented towards the rows of bases in reads and the consensus, then the slice view of an assembly is "vertical", meaning the assembly is oriented towards the tiling at each assembly position. With this picture in mind it is easy to understand that the contig view and the slice view is purely a shift in representation, and there is no change in information. In fact there are Slice Tools that perform the change in representation between contig and slice views of an assembly without losing or gaining information in the transformation.
Contig or Sequence View of an Assembly
"Horizontal Orientation"

The power of the slice format is that it makes operations that are difficult to perform within a contig representation trivial to perform with a slice representation. For example, recomputing the consensus of an assembly or merging two assemblies together are trivial because the alignment of the assembly is explicitly available without calculation in the slice view, and slices can be manipulated independently while maintaining the original relative tiling.
Slice View of an Assembly
"Vertical Orientation"
Back to top
Slice XML Format
The power of the Slice Tools come from their mutual ability to read and write Slice XML files. The Slice XML file format is a well defined structure for storing all of the elements of an assembly in a vertical orientation including the bases, quality values, consensus, position, and read information. The Slice Data can be quickly validated and parsed by a standard XML library, and then manipulated for the specific needs of the tool.
Example Assembly

The information for each slice is store within an XML Slice element. Adjacent Slice elements are stored within a SliceRange. The SliceRange allows for multiple assemblies to easily be stored within a single Slice XML file, much like how multiple assemblies can be stored within a single contig file.
Slice elements within a SliceRange:
<SliceRange Index="0" Gindex="0">
<Slice Index="0" Gindex="0" Existing="G">
<Nuc>GG</Nuc>
<Qualval>34 28</Qualval>
<ReadID>0 1</ReadID>
</Slice>
<Slice Index="1" Gindex="1" Existing="T">
<Nuc>TTTTT</Nuc>
<Qualval>28 30 36 32 33</Qualval>
<ReadID>0 1 2 3 4</ReadID>
</Slice>
<Slice Index="2" Gindex="2" Existing="G">
<Nuc>TGGG</Nuc>
<Qualval>21 27 13 36</Qualval>
<ReadID>1 2 3 4</ReadID>
</Slice>
<Slice Index="3" Gindex="3" Existing="T">
<Nuc>TT</Nuc>
<Qualval>17 37</Qualval>
<ReadID>3 4</ReadID>
</Slice>
</SliceRange>
|
From this example, we see the there is a small repeat of GTGT in the consensus from ungapped position 0 through ungapped position 3. In addition, the tiling information shows that there are 5 sequences (ReadIDs 0-4) that support this consensus at all 4 positions. The Slice Elements are contained within a SliceRange which shows the absolute coordinates of the slices. More information about the sequences themselves is available in the ReadCollection.
Read elements within a ReadCollection:
<ReadCollection>
<Read Id="0" Seqname="DMGLN85TF" Dir="0" Chem="">
<Clr Left="1" Right="2" />
</Read>
<Read Id="1" Seqname="DMGHB30TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
<Read Id="2" Seqname="DMGIC39TF" Dir="1" Chem="">
<Clr Left="2" Right="1" />
</Read>
<Read Id="3" Seqname="DMGRA39TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
<Read Id="4" Seqname="DMGNB33TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
</ReadCollection>
|
The Read Id list in the Slice elements index the full information about the sequence listed in the ReadCollection. This information is needed for quality class calculation and other operations. The standard Read can be augmented with the tool augmentSlice to include the bases and quality values not included in the tiling from outside of the clear range. When the Read elements are augmented with the trimmed sequences, the slice file contains the exact same information as the combination of the seq, qual, and contig files combined. Only one ReadCollection is allowed per Slice XML File.
A Slice XML file contains the read and slice information as above. A Slice XML file also requires the standard XML headers and a Request element which bundles the ReadCollection and SliceRange elements together into a complete and valid Slice XML file.
Complete Slice XML File
% cat example.slice
<?xml version="1.0" encoding="UTF-8"?>
<!DOCTYPE Request SYSTEM "http://slicetools.sourceforge.net/aserver.dtd">
<Request Type="Consensus" Host="getCoverage" User="getCoverage" Option="All">
<ReadCollection>
<Read Id="0" Seqname="DMGLN85TF" Dir="0" Chem="">
<Clr Left="1" Right="2" />
</Read>
<Read Id="1" Seqname="DMGHB30TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
<Read Id="2" Seqname="DMGIC39TF" Dir="1" Chem="">
<Clr Left="2" Right="1" />
</Read>
<Read Id="3" Seqname="DMGRA39TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
<Read Id="4" Seqname="DMGNB33TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
</ReadCollection>
<SliceRange Index="0" Gindex="0">
<Slice Index="0" Gindex="0" Existing="G">
<Nuc>GG</Nuc>
<Qualval>34 28</Qualval>
<ReadID>0 1</ReadID>
</Slice>
<Slice Index="1" Gindex="1" Existing="T">
<Nuc>TTTTT</Nuc>
<Qualval>28 30 36 32 33</Qualval>
<ReadID>0 1 2 3 4</ReadID>
</Slice>
<Slice Index="2" Gindex="2" Existing="G">
<Nuc>TGGG</Nuc>
<Qualval>21 27 13 36</Qualval>
<ReadID>1 2 3 4</ReadID>
</Slice>
<Slice Index="3" Gindex="3" Existing="T">
<Nuc>TT</Nuc>
<Qualval>17 37</Qualval>
<ReadID>3 4</ReadID>
</Slice>
</SliceRange>
</Request>
|
Note: | Many of the attributes and elements in a Slice XML file are optional. Please see the DTD for exact details. |
Back to top
Build Instructions
Prerequisites
Download the latest slicetools from the download page. After download, unpack the archive with tar:
$ tar xjvf slicetools-2.0.0.tar.bz2(the exact version number may change) Configure
Following this, you'll need to configure the make system. The first thing you have to decide is how to fetch the Slice XML DTD file. The DTD tells the XML processor how to test the Slice XML document is correct. It is essential that the DTD is accessible at all times, and from all machines that you intend to run the Slice Tools on. By default, the Slice Tools will request the DTD from the Slice Tools website (http://slicetools.sourceforge.net/aserver.dtd), but you may want to avoid this dependency and make the dtd available locally. If you decide you do want the DTD installed locally, you can specify either a URL or the exact file path to the dtd with the --with-slicedtd option. The build system will copy the dtd into the share directory of the installation.
After deciding how to fetch the DTD, run configure. If you installed Xerces in a non-system location, you'll also need to specify the prefix to the library and header files. This can be done with --with-xerces-prefix=PREFIX. Note you may need to adjust your LD_LIBRARY_PATH or other environment variables if libxerces-c.so is not in a system path.
At the University of Maryland, configure is run as:
$ ./configure --with-xercesc-prefix=/fs/sz-user-supported/`uname`-`uname -m`/ \
--with-slicedtd=/fs/sz-user-supported/share/dtd/ \
--prefix=/fs/sz-user-support/`uname`-`uname -m`
At the Institute for Genomic Research, configure is run as:
$ ./configure --with-xercesc-prefix=/usr/local/packages/Xerces \
--with-slicedtd=http://tools.tigr.org/dtds/aserver.dtd
Build
After configuration, you can then build and install the program.
$ make $ make install
'make install' will copy the tools into a bin directory within the build directory. It will also create a lib, include, and share directory with other supporting files. You may want to use the --prefix option with configure to specify a new path.
Back to top
Working with Slices
The choice of slice view or contig view of the assembly should be determined by the type of operations that will be performed. Operations which can be performed on a per assembly position basis such as computing the consensus, quality class, or average depth of coverage can immediately be computed from the slice view of the assembly. In addition, manipulation which join parts of assemblies, or split assemblies apart can usually be performed quicker and easier within a slice view.
A contig view of the assembly is appropriate for operations on sequences as a whole, such as assembling sequences together or computing average read length. Also, comparitive operations, such as aligning two sequencing, are typically done on the consensus of the contig. These alignments can be computed by converting the slices to fasta format with slice2fasta and then running an alignment program such as MUMmer.
Contig File of Example Assembly
% slice2contig example.slice
##0 5 4 bases, 00000000 checksum.
GTGT
#DMGLN85TF(0) [] 2 bases, 00000000 checksum. {1 2} <1 2>
GT
#DMGHB30TR(0) [RC] 3 bases, 00000000 checksum. {3 1} <1 3>
GTT
#DMGIC39TF(1) [RC] 2 bases, 00000000 checksum. {2 1} <2 3>
TG
#DMGRA39TR(1) [RC] 3 bases, 00000000 checksum. {3 1} <2 4>
TGT
#DMGNB33TR(1) [RC] 3 bases, 00000000 checksum. {3 1} <2 4>
TGT
|
In this simple example it is difficult to see if the alignment is homogenous, or what the quality value of the non-homogenous bases are without performing an alignment and without loading the quality values as well. In the Slice view above of the same assembly, it is immediately obvious that the slice with index 2 has a discrepant T with quality value of 21 with read id of 1.
As mentioned above, there are situations where the Slice view of the assembly is not appropriate or not possible, such as in manipulating sequences before assembly, so many of the Slice Tools were built to convert to and from the slice format. The pathways for converting assembly data are shown below.
Slice Format Transformations

Note: | The transformation performed by slice2contig only creates the contig file and does not generate the seq or qual files. |
The diagram above shows how to convert between the various formats. Central to all formats is a repository for the original read information. That information is typically stored in databases such as the trace archive. The unassembled sequences are assembled with a general use assembler such as Celera Assembler, or with the AMOS. If you assembly the sequences with minimus (part of AMOS), you can use bank2contig to convert the contigs to contig format. The contig files are then coverted to Slice format with getCoverage, where they can be manipulated with the slicetools. The updated slice contigs can then be converted back into regular contig format with slice2contig, or into an uploadable format with slice2tasm. See the tool descriptions below for more information.
Back to top
Consensus Calling
A number of the slice tools manipulate the contigs by either adding or removing reads from the contig. As a result the consensus at each position may need to be recalled. This operations is performed by the consensus calling routines available within libSlice which is a flexible C and C++ library for consensus calling and consensus quality calculation. More information is available on the libSlice website.
Back to top
Common Options
All of the Slice Tools support a standard set of Slice options beyond the common options provided by the Foundation classes for C++, ie. --debug, --help, --version. The common options and foundation API is documented on the libFoundation website.
Slice Options ------------- -s|--stats Display Statistics about processing -o|--output Specify output file (default to stdout) -c|--consensus Recall the consensus of each slice -e|--recall-empty Recall the consensus of empty slices (Containing no bases) -t|--highQuality |
Note: | Empty slices are slices that have a consensus, but no bases and no underlying sequences. They are usually the result of non-unique unitigs. In those slices the full tiling data is not available, so the Slice tools will not recall the consensus for those slices by default. |
More information on consensus calling and ambiguity codes is available on the libSlice website.
Back to top
addCoverage
Microassemble one or more query reads/assemblies onto a preexisting reference assembly. The reference assembly must provided in slice format (reference.slice). The query data must be provided in slice format, and be available within a directory specified by -s seqdir. The alignments for the query data is specified by a combination of a spec file and a snps file. This allows for complete control and flexibility for the user to choose the desired alignment. The spec file lists the query filename and alignment chain to use for assembling (one query per line). The snps file is a concatenation of the individual snps files for all query data (see zipclap for more information on creating a snps file). The alignments must be based on the consensus of the slice files provided (ie via slice2fasta), but the individual slice files can be single or multiple read assemblies. The program will error if you try to simultaneously add overlapping query data.
The reference assembly is first broken into pieces by splitSlice according to the alignment information for efficency. It then uses zipclap for merging the assemblies together. The alignment criterion of zipclap must therefore be satisfied, or the addCoverage opteration will fail. Finally the zipped components are merged together with mergeSlice into a single resultant slice file.
Usage: addCoverage.pl reference.slice reads.spec reads.snps [OPTIONS]
reference.slice Reference Assembly to add reads to
reads.spec File containing (seqname chain) pairs to add
reads.snps File containing snp data for all reads to add
options:
-s <seqdir> Specify directory that contains the read slice files
(DEFAULT: seqs)
-o <prefix> Specify prefix for result (DEFAULT: merge)
-w <workdir> Specify work directory (DEFAULT: work)
-[no]merge [Don't] merge the resultant slice file (DEFAULT: -merge)
-[no]recall [Don't] Recall the merged consensus (DEFAULT: -norecall)
-[no]contig [Don't] Convert result to contig (DEFAULT: -nocontig)
-[no]tcov [Don't] Convert result to tcov (DEFAULT: -notcov)
-restart Attempt to use intermediate files from previous run.
Default: erase previous work directory and restart from scratch
-0 Input slices are 0-based [Default]
-1 Input slices are 1-based
-v Toggle being more verbose
|
Example addCoverage Session
% grep '<Read ' reference.slice -c 457 % ls seqs/ BASC849TF.slice BARFC49TR.slice BARGW59T1GBA86F.slice BASER18TF.slice % cat spec.all BARFC49TR 0 BARGW59T1GBA86F 0 BASC849TF 0 BASER18TF 0 % cat reads.snps stripped.fasta, stripped, reads.fasta, BARGW59T1GBA86F, 0, 412, 1114, 1, 704, F, 457, 47, c, 2, I, ., c stripped.fasta, stripped, reads.fasta, BASER18TF, 0, 1224, 2025, 1, 800, F, 1949 , 725, a, 2, D, a, . stripped.fasta, stripped, reads.fasta, BASER18TF, 0, 1224, 2025, 1, 800, F, 1976 , 751, t, 2, D, t, . stripped.fasta, stripped, reads.fasta, BASC849TF, 0, 14666, 15416, 751, 1, R, 0, 0, ., 2, N, ., . stripped.fasta, stripped, reads.fasta, BARGW59T1GBA86FB, 0, 466, 1168, 1, 703, F , 0, 0, ., 2, N, ., . stripped.fasta, stripped, reads.fasta, BARFC49TR, 0, 35963, 36526, 1, 564, F, 0, 0, ., 2, N, ., . stripped.fasta, stripped, reads.fasta, BAREI33TR-1, 0, 1, 720, 1, 719, F, 711, 7 10, t, 2, D, t, . % addCoverage reference.slice spec.all reads.snps Loading Spec... Loading SNPs... Verifying Overlaps... Splitting... Zipping... 0 BARGW59T1GBA86F <412 1114> 1 BASER18TF <1224 2025> 2 BASC849TF <14666 15416> 3 BARFC49TR <35963 36526> Collecting Pieces... Merging... % grep '<Read ' merge.slice -c 461 |

Back to top
augmentSlice
Augments a Slice XML file with bases and quality values from outside of the CLR range. Bases and quality values outside the CLR range are added as TrimmedSeq records to each read in the ReadCollection.
Usage: augmentSlice [options] prefix slice.xml
prefix Prefix to .seq and .qual files
slice.xml Slice XML file
|
Example Read with TrimmedSeq Elements:
<Read Id="0" Seqname="DMGKS34TR" Dir="0" Chem="">
<Clr Left="27" Right="822" />
<TrimmedSeq position="left">
<Nuc>CGCTGTGCTGGAAAGACGTAATTTTA</Nuc>
<Qualval>13 07 12 11 07 09 09 18 19 17 17 09 07 07 13 08 07 07 11 17 16 09 07 08 09 12</Qualval>
</TrimmedSeq>
<TrimmedSeq position="right">
<Nuc>TGTATTTGCTTTTC</Nuc>
<Qualval>08 16 11 11 20 31 28 26 25 21 32 34 30 26</Qualval>
</TrimmedSeq>
</Read>
|
Back to top
getCoverage
getCoverage is not directly a Slice Tool because it does not operate from Slice XML files. Rather it is a tool that can convert from contig format to slice format by specifing the --xml flag to getCoverage. Ranges in the assembly can be specified if only a region of the assembly is of interest.
Usage: getCoverage [options] --xml prefix
prefix Prefix to .contig and .qual files
Standard Options
----------------
-a|--asmbl_id asmbl_id Specify single contig id in multi-contig file
(use 'all' for all contigs in file)
Coordinate Options
------------------
For XML, stats, and tiling modes, the default is to calculate coverage
information for every position in the contig. For all modes, a range of
positions (a cut) can be specified.
-x {--xg} pos Specify ungapped {gapped} position for start of cut
Negative values cut from position to end of contig
-y {--yg} pos Specify ungapped {gapped} position for end of cut
Negative values specify offset from -x pos or end of contig
-z|--radius size Specify a circular cut, use in conjunction with -x {--xg}
Other Options
-------------
-0 Use 0-based coordinates when displaying data (Default for XML)
-1 Use 1-based coordiantes when displaying data (Default for other modes)
--nogaps Display ungapped data only
--offset Generate read offset file for tiling mode
-G|--gapped Display gapped coordinates as well in tiling mode
-Q|--silent Do not display status messages
-R|--readid Show the Read index of each read in the idTbl in tiling mode
-S|--split Split output into separate files per contig (implies '-a all')
-o|--outdir path Specify output directory
-c mode Recall the consensus in specified ambiguity mode for stats file
(0=Off, 1=No ambiguity, 2=Minimal Ambiguity, 3=Annotation)
|
Example:
% getCoverage -l example.contig
<?xml version="1.0" encoding="UTF-8"?>
<!DOCTYPE Request SYSTEM "http://slicetools.sourceforge.net/aserver.dtd">
<Request Type="Consensus" Host="getCoverage" User="getCoverage" Option="All">
<ReadCollection>
<Read Id="0" Seqname="DMGLN85TF" Dir="0" Chem="">
<Clr Left="1" Right="2" />
</Read>
<Read Id="1" Seqname="DMGHB30TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
<Read Id="2" Seqname="DMGIC39TF" Dir="1" Chem="">
<Clr Left="2" Right="1" />
</Read>
<Read Id="3" Seqname="DMGRA39TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
<Read Id="4" Seqname="DMGNB33TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
</ReadCollection>
<SliceRange Index="0" Gindex="0">
<Slice Index="0" Gindex="0" Existing="G">
<Nuc>GG</Nuc>
<Qualval>34 28</Qualval>
<ReadID>0 1</ReadID>
</Slice>
<Slice Index="1" Gindex="1" Existing="T">
<Nuc>TTTTT</Nuc>
<Qualval>28 30 36 32 33</Qualval>
<ReadID>0 1 2 3 4</ReadID>
</Slice>
<Slice Index="2" Gindex="2" Existing="G">
<Nuc>TGGG</Nuc>
<Qualval>21 27 13 36</Qualval>
<ReadID>1 2 3 4</ReadID>
</Slice>
<Slice Index="3" Gindex="3" Existing="T">
<Nuc>TT</Nuc>
<Qualval>17 37</Qualval>
<ReadID>3 4</ReadID>
</Slice>
</SliceRange>
</Request>
|
getCoverage can also be used for creating tab deliminated or human readable views of an assembly and has an extremely flexible interface for specifying ranges of contigs to display. More information is available on the getCoverage page.
Back to top
mergeSlice
Merges multiple Slice XML files into a singe SliceRange within a single Slice file. The resultant ReadCollection will be renumbered and the the coordinates of the each slice will be adjusted.
Usage: mergeSlice [options] slice1.xml [slice2.xml [...]]
slice1.xml First Slice XML file
slice2.xml Second Slice XML file
Example Inputs:
--------------------------
slice1.xml slice2.xml
Slice 1 (A) Slice 1 (G)
Slice 2 (C) Slice 2 (T)
Example Output:
--------------------------
Slice 1 (A)
Slice 2 (C)
Slice 3 (G)
Slice 4 (T)
|
Note: |
mergeSlice does not merge individual Slice elements together, but rather
merges multiple SliceRange into a single SliceRange while
maintaining the original Slice elements. The size of the resulting
SliceRange will be the sum of the size of the input SliceRange
elements. The Slice Tool zipSlice is able to merge Slice elementes together by "zipping" two SliceRange elements into a single SliceRange that is at least as large as the larger of the two input SliceRange elements. |
Back to top
revSlice
Reverse compliments a slice XML file. The slices are written back in reverse order, and each base is reverse complimented. The read information is also reversed.
Usage: revSlice [options] slice.xml
slice.xml Slice XML file
|
Example:
% revSlice example.slice
<?xml version="1.0" encoding="UTF-8"?>
<!DOCTYPE Request SYSTEM "http://slicetools.sourceforge.net/aserver.dtd">
<Request Type="Consensus" Host="getCoverage" User="getCoverage" Option="All">
<ReadCollection>
<Read Id="0" Seqname="DMGLN85TF" Dir="1" Chem="">
<Clr Left="2" Right="1" />
</Read>
<Read Id="1" Seqname="DMGHB30TR" Dir="0" Chem="">
<Clr Left="1" Right="3" />
</Read>
<Read Id="2" Seqname="DMGIC39TF" Dir="0" Chem="">
<Clr Left="1" Right="2" />
</Read>
<Read Id="3" Seqname="DMGRA39TR" Dir="0" Chem="">
<Clr Left="1" Right="3" />
</Read>
<Read Id="4" Seqname="DMGNB33TR" Dir="0" Chem="">
<Clr Left="1" Right="3" />
</Read>
</ReadCollection>
<SliceRange Index="0" Gindex="0">
<Slice Index="0" Gindex="0" Existing="A">
<Nuc>AA</Nuc>
<Qualval>17 37</Qualval>
<ReadID>3 4</ReadID>
</Slice>
<Slice Index="1" Gindex="1" Existing="C">
<Nuc>ACCC</Nuc>
<Qualval>21 27 13 36</Qualval>
<ReadID>1 2 3 4</ReadID>
</Slice>
<Slice Index="2" Gindex="2" Existing="A">
<Nuc>AAAAA</Nuc>
<Qualval>28 30 36 32 33</Qualval>
<ReadID>0 1 2 3 4</ReadID>
</Slice>
<Slice Index="3" Gindex="3" Existing="C">
<Nuc>CC</Nuc>
<Qualval>34 28</Qualval>
<ReadID>0 1</ReadID>
</Slice>
</SliceRange>
</Request>
|
Back to top
rotateSlice
Rotates the tiling effectively moving the beginning of the consensus to the end. Since the count specifies the number of slices, all rotations are to the gapped consensus. The linear rotation is useful if you need to explicitly mark which reads cross the origin- the ReadCollection will contain new reads named read'. rotateSlice only performs counter-clockwise rotations, but you can simulate a clockwise rotation with a large CCW rotation.
rotateSlice Options ------------------ -n <count> Specify number of slices to rotate by -L Assume contig is linear, renumber rotated reads with read' |
Example:
% rotateSlice example.slice -n 1
<?xml version="1.0" encoding="UTF-8"?>
<!DOCTYPE Request SYSTEM "http://slicetools.sourceforge.net/aserver.dtd">
<Request Type="Consensus" Host="getCoverage" User="getCoverage" Option="All">
<ReadCollection>
<Read Id="0" Seqname="DMGLN85TF" Dir="0" Chem="">
<Clr Left="1" Right="2" />
</Read>
<Read Id="1" Seqname="DMGHB30TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
<Read Id="2" Seqname="DMGIC39TF" Dir="1" Chem="">
<Clr Left="2" Right="1" />
</Read>
<Read Id="3" Seqname="DMGRA39TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
<Read Id="4" Seqname="DMGNB33TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
</ReadCollection>
<SliceRange Index="0" Gindex="0">
<Slice Index="0" Gindex="0" Existing="T">
<Nuc>TTTTT</Nuc>
<Qualval>28 30 36 32 33</Qualval>
<ReadID>0 1 2 3 4</ReadID>
</Slice>
<Slice Index="1" Gindex="1" Existing="G">
<Nuc>TGGG</Nuc>
<Qualval>21 27 13 36</Qualval>
<ReadID>1 2 3 4</ReadID>
</Slice>
<Slice Index="2" Gindex="2" Existing="T">
<Nuc>TT</Nuc>
<Qualval>17 37</Qualval>
<ReadID>3 4</ReadID>
</Slice>
<Slice Index="3" Gindex="3" Existing="G">
<Nuc>GG</Nuc>
<Qualval>34 28</Qualval>
<ReadID>0 1</ReadID>
</Slice>
</SliceRange>
</Request>
|
Back to top
slice2contig
Converts a Slice XML file into contig format. By specifing the -C option, the contig is assumed circular, and there allows for reads to be present at both ends of the contig. In those cases, the reads will have a negative offset to indicate their tiling wraps the present origin. Unfortunately, reads with negative offsets cannot currently be processed with getCoverage.
Usage: slice2contig [options] slice.xml
slice.xml Slice XML file
slice2contig Options
--------------------
-i|--asmbl_id id Specify the id for the contig
-C Allow for circular contigs
(Some reads may have negative coordinates)
|
Example:
% slice2contig example.slice
##0 5 4 bases, 00000000 checksum.
GTGT
#DMGLN85TF(0) [] 2 bases, 00000000 checksum. {1 2} <1 2>
GT
#DMGHB30TR(0) [RC] 3 bases, 00000000 checksum. {3 1} <1 3>
GTT
#DMGIC39TF(1) [RC] 2 bases, 00000000 checksum. {2 1} <2 3>
TG
#DMGRA39TR(1) [RC] 3 bases, 00000000 checksum. {3 1} <2 4>
TGT
#DMGNB33TR(1) [RC] 3 bases, 00000000 checksum. {3 1} <2 4>
TGT
|
Note: | The transformation performed by slice2contig only creates the contig file and does not generate the seq or qual files. If those files are needed, first convert to tile with slice2tile, and then convert to contig with input_gen. |
Back to top
slice2fasta
Converts the ungapped consensus of a slice XML file into a fasta file.
Usage: slice2fasta [options] slice.xml
slice.xml Slice XML file
slice2fasta Options
-------------------
-i|--asmbl_id id Specify the id for the fasta record
|
Example:
% slice2fasta example.slice >0 5 4 bases, 00000000 checksum. GTGT |
Back to top
slice2tasm
Converts a slice XML file into a tasm file suitable for upload.
Usage: slice2tasm [options] slice.xml
slice.xml Slice XML file
slice2tasm Options
-------------------
-i|--asmbl_id <id> Specify the id for the tasm record
-I|--ca_id <id> Specify the ca_contig_id
-d|--method <method> Specify the method string
-n|--comment <comment> Specify the comment string
-u|--user <username> Specify the userid
-T|--date <datestring> Specify the mod_date and ed_date
|
Example:
% slice2tasm example.slice sequence GTGT lsequence GTGT quality 0x02020803 asmbl_id 0 seq_id com_name type method slice2tasm ed_status redundancy 3.00 perc_N 0.00 seq# 5 full_cds cds_start cds_end ed_pn mschatz ed_date 09/12/03 01:01:23 PM mod_date 09/12/03 01:01:23 PM comment CA FREE: 0 frameshift seq_name DMGLN85TF asm_lend 1 asm_rend 2 seq_lend 1 seq_rend 2 best comment db offset 0 lsequence GT seq_name DMGHB30TR asm_lend 1 asm_rend 3 seq_lend 3 seq_rend 1 best comment db offset 0 lsequence GTT seq_name DMGIC39TF asm_lend 2 asm_rend 3 seq_lend 2 seq_rend 1 best comment db offset 1 lsequence TG seq_name DMGRA39TR asm_lend 2 asm_rend 4 seq_lend 3 seq_rend 1 best comment db offset 1 lsequence TGT seq_name DMGNB33TR asm_lend 2 asm_rend 4 seq_lend 3 seq_rend 1 best comment db offset 1 lsequence TGT |
Back to top
slice2tile
Converts a slice XML file into tile XML format suitable for input_gen.
Usage: slice2tile [options] slice.xml
slice.xml Slice XML file
|
Example:
% slice2tile example.slice
<?xml version="1.0" encoding="UTF-8"?>
<!DOCTYPE Request SYSTEM "http://slicetools.sourceforge.net/aserver.dtd">
<Request Type="Assembly" Host="slice2tile" User="slice2tile">
<Source Project="default" Number="default" BacID="0" ID="1" Server="default" />
<Assembly>
<SequenceSet>
<Sequence Id="0" Name="DMGLN85TF">
<Nuc>GT</Nuc>
<Qualval>34 28</Qualval>
<Clr Left="1" Right="2" />
</Sequence>
<Sequence Id="1" Name="DMGHB30TR">
<Nuc>AAC</Nuc>
<Qualval>21 30 28</Qualval>
<Clr Left="1" Right="3" />
</Sequence>
<Sequence Id="2" Name="DMGIC39TF">
<Nuc>CA</Nuc>
<Qualval>27 36</Qualval>
<Clr Left="1" Right="2" />
</Sequence>
<Sequence Id="3" Name="DMGRA39TR">
<Nuc>ACA</Nuc>
<Qualval>17 13 32</Qualval>
<Clr Left="1" Right="3" />
</Sequence>
<Sequence Id="4" Name="DMGNB33TR">
<Nuc>ACA</Nuc>
<Qualval>37 36 33</Qualval>
<Clr Left="1" Right="3" />
</Sequence>
</SequenceSet>
<LibrarySet />
<Links />
<ContigSet>
<Contig Id="ASM_0">
<Nuc>GTGT</Nuc>
<Seq Id="0">
<Gaps />
<Seqrange Left="1" Right="2" />
<Asmrange Left="1" Right="2" />
<Offset>0</Offset>
</Seq>
<Seq Id="1">
<Gaps />
<Seqrange Left="3" Right="1" />
<Asmrange Left="1" Right="3" />
<Offset>0</Offset>
</Seq>
<Seq Id="2">
<Gaps />
<Seqrange Left="2" Right="1" />
<Asmrange Left="2" Right="3" />
<Offset>1</Offset>
</Seq>
<Seq Id="3">
<Gaps />
<Seqrange Left="3" Right="1" />
<Asmrange Left="2" Right="4" />
<Offset>1</Offset>
</Seq>
<Seq Id="4">
<Gaps />
<Seqrange Left="3" Right="1" />
<Asmrange Left="2" Right="4" />
<Offset>2</Offset>
</Seq>
</Contig>
</ContigSet>
</Assembly>
</Request>
|
Back to top
splitSlice
Slice a slice file into multiple slice files based on a list of partition points
Usage: splitSlice [splitSlice Options] slice.xml
slice.xml Slice XML file
splitSlice Options
------------------
-p <comma separated partions> List of slices to partition on (index)
-f <filename> File containing list of slices to partition on (index)
|
Example:
% splitSlice -p 1,3 example.slice % grep '<Slice' example_*.slice example_0.slice: <SliceRange Index="0" Gindex="0"> example_0.slice: <Slice Index="0" Gindex="0" Existing="G"> example_1.slice: <SliceRange Index="1" Gindex="1"> example_1.slice: <Slice Index="1" Gindex="1" Existing="T"> example_1.slice: <Slice Index="2" Gindex="2" Existing="G"> example_2.slice: <SliceRange Index="3" Gindex="3"> example_2.slice: <Slice Index="3" Gindex="3" Existing="T"> |
Back to top
SliceService
Recalls the consensus and calculates quality classes or consensus quality values. By default, the SliceService recalls the consensus and calculates the quality class of the slices. Specifying any of -v, -q, -c, or their associated SimpleOptions will cause SliceService to perform only what is specified.
Output is into a raw Consensus block which needs an XML header and to be put into a Response block to be a valid XML file.
The default ambiguity mode is 3 (Conic Model).
Usage: SliceService [options] slice.xml
slice.xml Slice XML file
SliceService Options
------------------
-v|--qualityvalue Calculate the consensus quality value
-q|--qualityclass Calculate the quality class
-c|--consensus Recall the consensus (Same as the Slice Option)
|
Many of the SliceService Options can also be controlled by setting SimpleOptions in the slice file. The options are encoded in simple name and value pairs. SimpleOptions override command line options.
Understood SimpleOptions
| SimpleOption Name | Understood Values | Meaning |
|---|---|---|
| AmbiguityMethod | "None" or "0" | Recall the consensus with no ambiguity codes |
| "Minimal" or "1" | Recall the consensus with the minimal ambiguity model (Churchill & Waterman) |
|
| "Annotation" or "2" | Recall the consensus with the annotation ambiguity model | "Conic" or "3" | Recall the consensus with the conic ambiguity model |
| HighQualityThreshold | "30" (Any integer value) |
Set the high quality threshold |
| DoConsensus | "1" or "0" | Enables or Disables returning the consensus |
| DoQualityClasses | "1" or "0" | Enables or Disables returning the quality classes |
| DoQualityValues | "1" or "0" | Enables or Disables returning the consensus quality values |
The default options for the SliceService can be encoded as:
<?xml version="1.0" encoding="UTF-8"?>
<!DOCTYPE Request SYSTEM "http://slicetools.sourceforge.net/aserver.dtd">
<Request Type="Consensus" Host="getCoverage" User="getCoverage" Option="All">
<OptionBlock>
<SimpleOption Name="HighQualityThreshold" Value="30" />
<SimpleOption Name="AmbiguityMethod" Value="Annotation" />
<SimpleOption Name="DoConsensus" Value="1"/>
<SimpleOption Name="DoQualityClasses" Value="1">
<SimpleOption Name="DoQualityValues" Value="0"/>
</OptionBlock>
<ReadCollection>
<Read Id="0" Seqname="DMGLN85TF" Dir="0" Chem="" />
</ReadCollection>
<SliceRange>
<Slice>
<Nuc>G</Nuc>
<Qualval>26</Qualval>
<ReadID>0</ReadID>
</Slice>
</SliceRange>
</Request>
|
Example:
% SliceService example.slice <Consensus> <Nuc>GTGT</Nuc> <Qualclass>2 2 8 3</Qualclass> </Consensus> |
More information on consensus calling and ambiguity codes is available at http://intranet/software_docs/libSlice/.
More information is available on the aserver and the SliceService at http://intranet/ifx/closure/projects/assembly_server/index.shtml.
Back to top
stripSlice
Strip one or more Read elements from the tiling in Slice XML format.
Usage: stripSlice [options] slice.xml
slice.xml Slice XML file
stripSlice Options
------------------
-i|--id id Specify single read id to strip
-I|--idlist file Specify file of read ids to strip
-n|--seqname name Specify single seqname to strip
-N|--seqlist file Specify file of seqnames to strip
-r|--renumber Renumber read ids after stripping
|
Example:
% stripSlice -i 1 example.slice
<?xml version="1.0" encoding="UTF-8"?>
<!DOCTYPE Request SYSTEM "http://slicetools.sourceforge.net/aserver.dtd">
<Request Type="Consensus" Host="getCoverage" User="getCoverage" Option="All">
<ReadCollection>
<Read Id="0" Seqname="DMGLN85TF" Dir="0" Chem="">
<Clr Left="1" Right="2" />
</Read>
<Read Id="2" Seqname="DMGIC39TF" Dir="1" Chem="">
<Clr Left="2" Right="1" />
</Read>
<Read Id="3" Seqname="DMGRA39TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
<Read Id="4" Seqname="DMGNB33TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
</ReadCollection>
<SliceRange Index="0" Gindex="0">
<Slice Index="0" Gindex="0" Existing="G">
<Nuc>G</Nuc>
<Qualval>34</Qualval>
<ReadID>0</ReadID>
</Slice>
<Slice Index="1" Gindex="1" Existing="T">
<Nuc>TTTT</Nuc>
<Qualval>28 36 32 33</Qualval>
<ReadID>0 2 3 4</ReadID>
</Slice>
<Slice Index="2" Gindex="2" Existing="G">
<Nuc>GGG</Nuc>
<Qualval>27 13 36</Qualval>
<ReadID>2 3 4</ReadID>
</Slice>
<Slice Index="3" Gindex="3" Existing="T">
<Nuc>TT</Nuc>
<Qualval>17 37</Qualval>
<ReadID>3 4</ReadID>
</Slice>
</SliceRange>
</Request>
|
Back to top
trSlice
Translates the coordinates of Slice and SliceRange elements to new values. trSlice can also be used to quickly recall the consensus on slices.
Usage: trSlice [options] slice.xml
slice.xml Slice XML file
trSlice Options
------------------
-i|--index Set the new Slice Index start
-g|--gindex Set the new Slice Gindex start
-x|--sindex Set the new SliceRange Index
-y|--sgindex Set the new SliceRange Gindex
|
Example:
% trSlice -i 1 -g 2 -x 3 -y 4 example.slice
<?xml version="1.0" encoding="UTF-8"?>
<!DOCTYPE Request SYSTEM "http://slicetools.sourceforge.net/aserver.dtd">
<Request Type="Consensus" Host="getCoverage" User="getCoverage" Option="All">
<ReadCollection>
<Read Id="0" Seqname="DMGLN85TF" Dir="0" Chem="">
<Clr Left="1" Right="2" />
</Read>
<Read Id="1" Seqname="DMGHB30TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
<Read Id="2" Seqname="DMGIC39TF" Dir="1" Chem="">
<Clr Left="2" Right="1" />
</Read>
<Read Id="3" Seqname="DMGRA39TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
<Read Id="4" Seqname="DMGNB33TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
</ReadCollection>
<SliceRange Index="3" Gindex="4">
<Slice Index="1" Gindex="2" Existing="G">
<Nuc>GG</Nuc>
<Qualval>34 28</Qualval>
<ReadID>0 1</ReadID>
</Slice>
<Slice Index="2" Gindex="3" Existing="T">
<Nuc>TTTTT</Nuc>
<Qualval>28 30 36 32 33</Qualval>
<ReadID>0 1 2 3 4</ReadID>
</Slice>
<Slice Index="3" Gindex="4" Existing="G">
<Nuc>TGGG</Nuc>
<Qualval>21 27 13 36</Qualval>
<ReadID>1 2 3 4</ReadID>
</Slice>
<Slice Index="4" Gindex="5" Existing="T">
<Nuc>TT</Nuc>
<Qualval>17 37</Qualval>
<ReadID>3 4</ReadID>
</Slice>
</SliceRange>
</Request>
|
Back to top
zipSlice
Zip together a query slice file onto a reference slice file. The beginning of the alignment is specified with -O, and gaps can be inserted into both slice files using -r and -q. There is no restriction on how the slices (assemblies) overlap, so zipSlice can be used to perform exotic contig jumpstarting or other MicroAssembler operations.
zipSlice Options
--------------------
-R Query should be reversed before merging
-1 Coordinates of gaps are 1-based
-U Coordinates of gaps to insert are ungapped
-p Automatically promote preexisting gaps
--refcons Use the consensus of the reference for merged slices
(Uses the query consensus by default)
-q <csl> Comma separated list of positions to insert gaps in the query
-r <csl> Comma separated list of positions to insert gaps in the reference
-O <offset> Specify alignment offset from query to reference
(negative means reference left flanks query (always 0-based))
|
Example:
% cat read.slice
<?xml version="1.0" encoding="UTF-8"?>
<!DOCTYPE Request SYSTEM "http://slicetools.sourceforge.net/aserver.dtd">
<Request Type="Consensus" Host="getCoverage" User="getCoverage" Option="All">
<ReadCollection>
<Read Id="0" Seqname="DMGLN85TFB" Dir="0" Chem="">
<Clr Left="1" Right="4" />
</Read>
</ReadCollection>
<SliceRange Index="0" Gindex="0">
<Slice Index="0" Gindex="0" Existing="G">
<Nuc>G</Nuc>
<Qualval>34</Qualval>
<ReadID>0</ReadID>
</Slice>
<Slice Index="1" Gindex="1" Existing="A">
<Nuc>A</Nuc>
<Qualval>28</Qualval>
<ReadID>0</ReadID>
</Slice>
<Slice Index="2" Gindex="2" Existing="G">
<Nuc>G</Nuc>
<Qualval>21</Qualval>
<ReadID>0</ReadID>
</Slice>
<Slice Index="3" Gindex="3" Existing="T">
<Nuc>T</Nuc>
<Qualval>24</Qualval>
<ReadID>0</ReadID>
</Slice>
</SliceRange>
</Request>
% zipSlice read.slice example.slice -O 2
<?xml version="1.0" encoding="UTF-8"?>
<!DOCTYPE Request SYSTEM "http://slicetools.sourceforge.net/aserver.dtd">
<Request Type="Consensus" Host="getCoverage" User="getCoverage" Option="All">
<ReadCollection>
<Read Id="0" Seqname="DMGLN85TFB" Dir="0" Chem="">
<Clr Left="1" Right="4" />
</Read>
<Read Id="1" Seqname="DMGLN85TF" Dir="0" Chem="">
<Clr Left="1" Right="2" />
</Read>
<Read Id="2" Seqname="DMGHB30TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
<Read Id="3" Seqname="DMGIC39TF" Dir="1" Chem="">
<Clr Left="2" Right="1" />
</Read>
<Read Id="4" Seqname="DMGRA39TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
<Read Id="5" Seqname="DMGNB33TR" Dir="1" Chem="">
<Clr Left="3" Right="1" />
</Read>
</ReadCollection>
<SliceRange Index="0" Gindex="0">
<Slice Index="0" Gindex="0" Existing="G">
<Nuc>G</Nuc>
<Qualval>34</Qualval>
<ReadID>0</ReadID>
</Slice>
<Slice Index="1" Gindex="1" Existing="A">
<Nuc>A</Nuc>
<Qualval>28</Qualval>
<ReadID>0</ReadID>
</Slice>
<Slice Index="2" Gindex="2" Existing="G">
<Nuc>GGG</Nuc>
<Qualval>21 34 28</Qualval>
<ReadID>0 1 2</ReadID>
</Slice>
<Slice Index="3" Gindex="3" Existing="T">
<Nuc>TTTTTT</Nuc>
<Qualval>24 28 30 36 32 33</Qualval>
<ReadID>0 1 2 3 4 5</ReadID>
</Slice>
<Slice Index="4" Gindex="4" Existing="G">
<Nuc>TGGG</Nuc>
<Qualval>21 27 13 36</Qualval>
<ReadID>2 3 4 5</ReadID>
</Slice>
<Slice Index="5" Gindex="5" Existing="T">
<Nuc>TT</Nuc>
<Qualval>17 37</Qualval>
<ReadID>4 5</ReadID>
</Slice>
</SliceRange>
</Request>
|
Back to top
zipclap
zipclap is a front end for zipSlice for merging assemblies together. It is not exclusively a Slice Tool, because it also allows for contig file inputs in addition to slice file inputs. It can encapsulate performing the nucmer alignment between the assemblies, but can also be explicitly provided the alignment information. zipclap can be used to zip any pair of contigs together- including contigs made of just single reads. Since it uses nucmer to determine alignments it allows for extreme flexibility in allowing the operator to decide how and where assemblies should be merged. The desired alignment can be selected by passing the chain number (starting at 0) with the -c option, otherwise the first chain (chain 0) will be used.
Unlike zipSlice, zipClap requires that the alignment range is maximal, so that a proper overlap between the slices is provided. In the case where a non-maximal alignment is provided, and there are unaligned bases that would otherwise be forced together, zipSlice will error. See zipclaptypes.pdf for the details of the enforcement.
Usage: zipclap reference query [options]
reference and query can be either .contig or .slice files.
Options:
--------
-c <chain> specify alignment chain to use (DEFAULT 0)
-r <refshift> specify reference shift (DEFAULT 0)
-C Assume contig to be circular when converting to contig
-[no]contig [Don't] Create a contig file of result (DEFAULT: -contig)
-[no]tcov [Don't] Create a tcov file of result (DEFAULT: -tcov)
-[no]tasm [Don't] Create a tasm file of result (DEFAULT: -notasm)
-[no]fasta [Don't] Create a fasta file of result (DEFAULT: -nofasta)
-o <prefix> Specify prefix for out files (DEFAULT: zip)
-q <filename> Override snps file query fasta filename
-v Toggle being more verbose
The reference assembly is held "in place" and the query may be reverse
complemented or shifted until it aligns well. Alignment gaps are determined
by the snps file (ultimately via nucmer) and ensure the assemblies are aligned
properly.
Alignment information is searched in the following order so that if you
provide a snps file, then nothing else will be searched.
1) outprefix.snps
2) outprefix.aligns
3) outprefix.delta
4) nucmer
|
Example:
% head -1 a.contig ##a 1 749 bases, 73072043 checksum. % head -1 d.contig ##d 1 748 bases, D45CB0F5 checksum. % zipclap a.contig d.contig -o a-d -v Reference prefix is "a" Query prefix is "d" Getting fasta id from a.fasta... a Getting fasta id from d.fasta... d nucmer a d... ok. Loading a-d.snps Type III. qalignstart 1 ralignstart 1 offset 0 (xxdiff d.tcov a-d.tcov) Insert 1 gaps in query at 747 zipSlice... ok. % head -1 a-d.contig ##a-d 2 749 bases, 00000000 checksum. |
Back to top
Acknowledgements and History
Michael Schatz is the principal author of the Slice Tools package and conceived of many ideas including the conic ambiguity consensus caller. The getCoverage program was written by Pawel Gajer and engineered by Michael Schatz. Components and auxiliary scripts delivered with this package were written by Erik Ferlanti, Pawel Gajer, Daniel Kosack, Miguel Covarrubias, Adam Phillippy, and Martin Shumway. Early adopters who provided usage and feedback include Abhilasha Chaudhary, Mihai Pop, Seth Schobel, and Jeff Sitz. Evaluation of the universal consensus caller was done by an expert panel that included Robert Fleischmann, Herve Tettelin, Luke Tallon, and Jessica Vamathevan. This work was supported in part by the NIAID Bioinformatics Resource Center contract.
The Slice Tools were developed at the Bioinformatics Department of The Institute for Genomic Research (TIGR) under the direction of Steven Salzberg. The initial idea of representing multiple alignments in vertical form arose from work TIGR did on the Florida Anthrax attack in 2001-2002, and was motivated by the need to quantitatively assess single nucleotide polymophisms (SNPs). A prototype version of the Microassembler technology was written in perl by Martin Shumway and included an implementation of a gap promotion algorithm. This was used to electronically finish the B. anthracis Ames Ancestor reference genome. The Slice Tools technology has been in production at TIGR since 2003 and provides a comprehensive multiple alignment processing workbench and a universal consensus caller for genome assembly, editing, finishing, and archival. Slice Tools are instrumental in higher level assembly and finishing tools, including the Influenza sequencing assembly pipeline, AutoJoiner (a finishing tool for automatically closing sequencing gaps, link coming soon), and software to populate the NIAID Trace Assembly Archive.
Back to top
Support Information
Please visit the Slice Tools page at Sourceforge (http://www.sourceforge.net/projects/slicetools) or post on the slice tools help mailing list (slicetools-help [a t] lists . sourceforge . net).
Back to top